
Data Science Project Management Concepts
Data Science & Project Management Concepts
Tools of trade
Github for versioning.
- README in directories
- Trello/github
- /project – anal1 …
- /data – raw, unprocessed data
- /refererences – fastq files and human references
- /code – Javabased or compiled
- ~/bin – where executables go
- ~/scripts – where scripts go
- .bashrc settings for bash login
Early. Prototype the entire pipeline or analysis before cleaning up or creating elegant code.
Second. Create unit tests for each step
Third – only if it is going to be used. Remove hardcoding and add elegant solutions iteratively.
Waterfall vs Agile
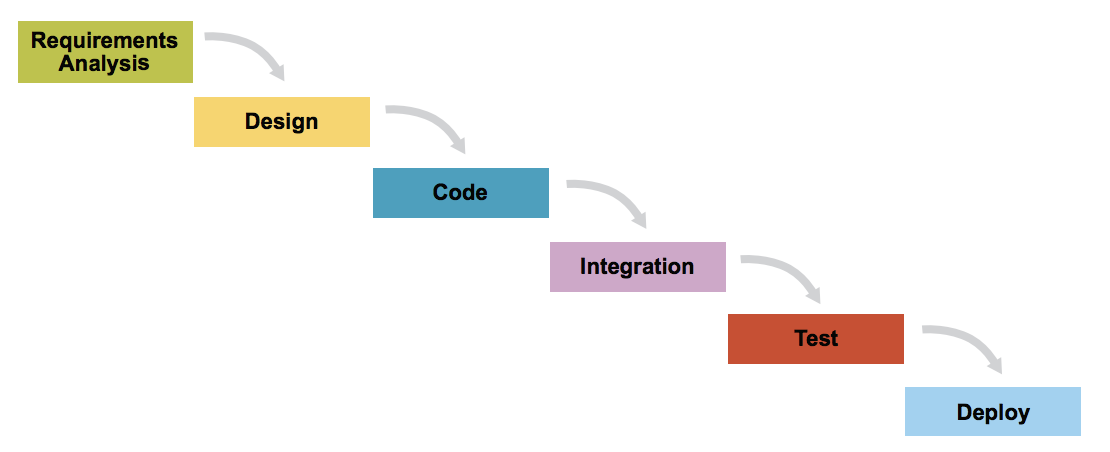
Waterfall. waterfall model is a relatively linear sequential design approach for certain areas of engineering design. In software development, it tends to be among the less iterative and flexible approaches, as progress flows in largely one direction (“downwards” like a waterfall) through the phases of conception, initiation, analysis, design, construction, testing, deployment and maintenance.
The waterfall development model originated in the manufacturing and construction industries; where the highly structured physical environments meant that design changes became prohibitively expensive much sooner in the development process. When first adopted for software development, there were no recognized alternatives for knowledge-based creative work.
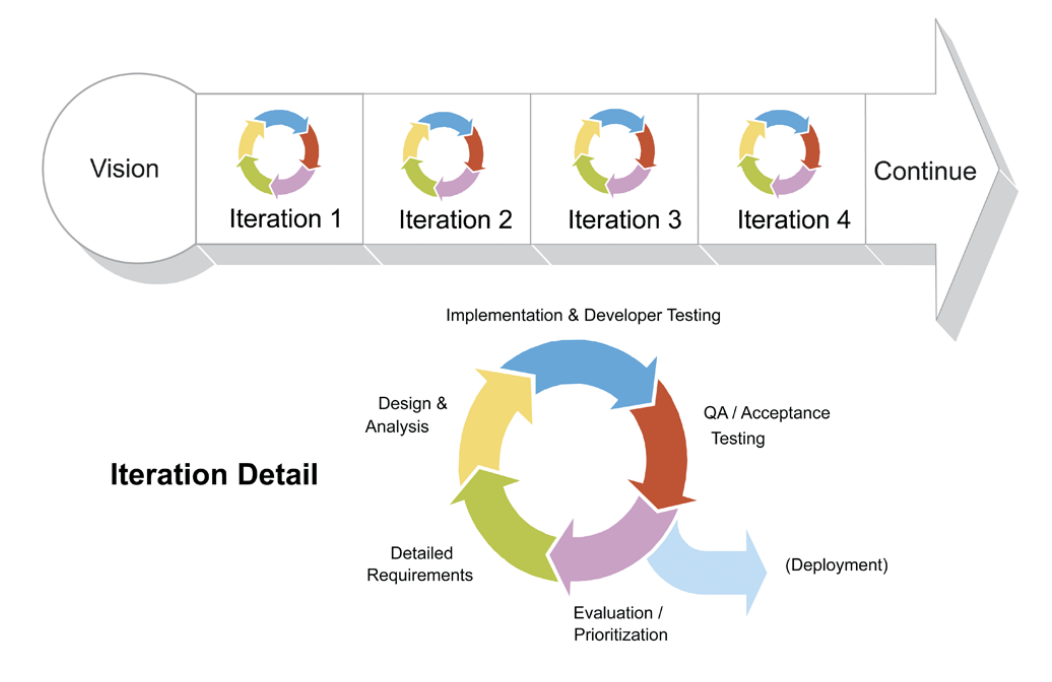
Agile software development is based on an incremental, iterative approach. Instead of in-depth planning at the beginning of the project, Agile methodologies are open to changing requirements over time and encourages constant feedback from the end-users. Cross-functional teams work on iterations of a product over a period of time, and this work is organized into a backlog that is prioritized based on business or customer value. The goal of each iteration is to produce a working product. In Agile methodologies, leadership encourages teamwork, accountability, and face-to-face communication. Business stakeholders and developers must work together to align the product with customer needs and company goals. Agile refers to any process that aligns with the concepts of the Agile Manifesto. In February 2001, 17 software developers met in Utah to discuss lightweight development methods. They published the Manifesto for Agile Software Development, which covered how they found “better ways of developing software by doing it and helping others do it” and included four values and 12 principles.
Waterfall
Agile
Manifesto for Agile Software Development
We are uncovering better ways of developing software by doing it and helping others do it. Through this work we have come to value: Individuals and interactions over processes and tools Working software over comprehensive documentation Customer collaboration over contract negotiation Responding to change over following a plan That is, while there is value in the items on the right, we value the items on the left more.
The fable of the Chicken and the Pig is used to illustrate the different levels of project stakeholders involved in a project. The basic fable runs:
-
- A Pig and a Chicken are walking down the road.
- The Chicken says: “Hey Pig, I was thinking we should open a restaurant!”
- Pig replies: “Hm, maybe, what would we call it?”
- The Chicken responds: “How about ‘ham-n-eggs’?”
- The Pig thinks for a moment and says: “No thanks. I’d be committed, but you’d only be involved.”
Sometimes, the story is presented as a riddle
-
- Question: In a bacon-and-egg breakfast, what’s the difference between the Chicken and the Pig?
- Answer: The Chicken is involved, but the Pig is committed!
In the last few years as agile has been rolled out to larger organizations, a new role has been identified. We call this the rooster. The rooster is very loud, disconnected from the work, and rarely knows what they are talking about.
Agile software development principles
The Manifesto for Agile Software Development is based on twelve principles:
- Customer satisfaction by early and continuous delivery of valuable software
- Welcome changing requirements, even in late development
- Working software is delivered frequently (weeks rather than months)
- Close, daily cooperation between business people and developers
- Projects are built around motivated individuals, who should be trusted
- A face-to-face conversation is the best form of communication (co-location)
- Working software is the primary measure of progress
- Sustainable development, able to maintain a constant pace
- Continuous attention to technical excellence and good design
- Simplicity—the art of maximizing the amount of work not done—is essential
- Best architectures, requirements, and designs emerge from self-organizing teams
- Regularly, the team reflects on how to become more effective, and adjusts accordingly
Iterative, incremental and evolutionary
Most agile development methods break product development work into small increments that minimize the amount of up-front planning and design. Iterations, or sprints, are short time frames (timeboxes) that typically last from one to four weeks. Each iteration involves a cross-functional team working in all functions: planning, analysis, design, coding, unit testing, and acceptance testing. At the end of the iteration, a working product is demonstrated to stakeholders. This minimizes the overall risk and allows the product to adapt to changes quickly. An iteration might not add enough functionality to warrant a market release, but the goal is to have an available release (with minimal bugs) at the end of each iteration.[23] Multiple iterations might be required to release a product or new features.
Working software is the primary measure of progress.
Efficient and face-to-face communication
No matter which development method is followed, every team should include a customer representative (Product Owner in Scrum). This person is agreed by stakeholders to act on their behalf and makes a personal commitment to being available for developers to answer questions throughout the iteration. At the end of each iteration, stakeholders and the customer representative review progress and re-evaluate priorities with a view to optimizing the return on investment (ROI) and ensuring alignment with customer needs and company goals.
In agile software development, an information radiator is a (normally large) physical display located prominently near the development team, where passers-by can see it. It presents an up-to-date summary of the product development status. A build light indicator may also be used to inform a team about the current status of their product development.
Very short feedback loop and adaptation cycle
A common characteristic in agile software development is the daily stand-up (also known as the daily scrum). In a brief session, team members report to each other what they did the previous day toward their team’s iteration goal, what they intend to do today toward the goal, and any roadblocks or impediments they can see to the goal.[26]
Quality focus. Specific tools and techniques, such as continuous integration, automated unit testing, pair programming, test-driven development, design patterns, behavior-driven development, domain-driven design, code refactoring, and other techniques are often used to improve quality and enhance product development agility.[27] The idea is that the quality is built into the software. [28]
Scrum
Differences and Similarities Between Agile and Scrum
While Agile and Scrum follow the same system, there are some differences when comparing Scrum vs Agile. Agile describes a set of principles in the Agile Manifesto for building software through iterative development. On the other hand, Scrum is a specific set of rules to follow when practicing Agile software development. Agile is the philosophy and Scrum is the methodology to implement the Agile philosophy.
Because Scrum is one way to implement Agile, they both share many similarities. They both focus on delivering software early and often, are iterative processes, and accommodate change. They also encourage transparency and continuous improvement.
Adapted from Michael James and Luke Walter for CollabNet, Inc
Product Owner.
- Single person responsible for maximizing the return on investment (ROI) of the development effort
- Responsible for product vision
- Constantly re-prioritizes the Product Backlog, adjusting any longterm expectations such as release plans
- Final arbiter of requirements questions • Decides whether to release • Decides whether to continue development
- Considers stakeholder interests
- May contribute as a team member
- Has a leadership role
Scrum Master
- Works with the organization to make Scrum possible •
- Ensures Scrum is understood and enacted
- Creates an environment conducive to team self-organization •
- Shields the team from external interference and distractions to keep it in group flow (a.k.a. the zone) •
- Promotes improved engineering practices • Has no management authority over the team •
- Helps resolve impediments •
- Has a leadership role
Sprint Planning Meeting. At the beginning of each Sprint, the Product Owner and team hold a Sprint Planning Meeting to negotiate which Product Backlog Items they will attempt to convert to the working product during the Sprint. The Product Owner is responsible for declaring which items are the most important to the business. The Development Team is responsible for selecting the amount of work they feel they can implement without accruing technical debt. The team “pulls” work from the Product Backlog to the Sprint Backlog.
Daily Scrum and Sprint methods Every day at the same time and place, the Scrum Development Team members spend a total of 15 minutes inspecting their progress toward the Sprint goal, and creating a plan for the day. Team members share with each other what they did the previous day to help meet the Sprint goal, what they’ll do today, and what impediments they face.1
Extreme programming (XP)
Extreme Programming (XP) is an agile software development framework that aims to produce higher quality software and higher quality of life for the development team. XP is the most specific of the agile frameworks regarding appropriate engineering practices for software development.
The five values of XP are communication, simplicity, feedback, courage, and respect and are described in more detail below.
Communication. Software development is inherently a team sport that relies on communication to transfer knowledge from one team member to everyone else on the team. XP stresses the importance of the appropriate kind of communication – face to face discussion with the aid of a whiteboard or another drawing mechanism.
Simplicity. Simplicity means “what is the simplest thing that will work?” The purpose of this is to avoid waste and do only absolutely necessary things such as keep the design of the system as simple as possible so that it is easier to maintain, support, and revise. Simplicity also means address only the requirements that you know about; don’t try to predict the future.
Feedback. Through constant feedback about their previous efforts, teams can identify areas for improvement and revise their practices. Feedback also supports simple design. Your team builds something, gathers feedback on your design and implementation, and then adjust your product going forward.
Courage. KCourage as “effective action in the face of fear” (Extreme Programming Explained P. 20). This definition shows a preference for action based on other principles so that the results aren’t harmful to the team. You need courage to raise organizational issues that reduce your team’s effectiveness. You need courage to stop doing something that doesn’t work and try something else. You need courage to accept and act on feedback, even when it’s difficult to accept.
Respect. The members of your team need to respect each other in order to communicate with each other, provide and accept feedback that honors your relationship, and to work together to identify simple designs and solutions.
Whole Team. A cross functional group of people with the necessary roles for a product form a single team. This means people with a need as well as all the people who play some part in satisfying that need all work together on a daily basis to accomplish a specific outcome.
Informative Workspace. Set up your team space to facilitate face to face communication, allow people to have some privacy when they need it, and make the work of the team transparent to each other and to interested parties outside the team. Utilize Information Radiators to actively communicate up-to-date information.
Pair Programming. Pair Programming means all production software is developed by two people sitting at the same machine. The idea behind this practice is that two brains and four eyes are better than one brain and two eyes. You effectively get a continuous code review and quicker response to nagging problems that may stop one person dead in their tracks.
Teams that have used pair programming have found that it improves quality and does not actually take twice as long because they are able to work through problems quicker and they stay more focused on the task at hand, thereby creating less code to accomplish the same thing.
Kanban
Kanban is a scheduling system for lean manufacturing and just-in-time manufacturing (JIT). Taiichi Ohno, an industrial engineer at Toyota, developed kanban to improve manufacturing efficiency. Kanban is one method to achieve JIT.[3] The system takes its name from the cards that track production within a factory. For many in the automotive sector Kanban is known as “Toyota nameplate system” and as such, some other automakers won’t use the term Kanban.
Content sources include https://www.wikipedia.org/ and are available following Commons sharing.
GPL is a copyleft license essentially making it impossible to use code if one isn’t going to distribute it using the same license.
For this course, we will generally use the MIT License.
Permissive
BSD License
The BSD license is a simple license that merely requires that all code licensed under the BSD license be licensed under the BSD license if redistributed in source code format. BSD (unlike some other licenses) does not require that source code be distributed at all. “here’s the source code, do whatever you want with it, but if you have problems, it’s your problem”
MIT
Conditions only requiring preservation of copyright and license notices. Licensed works, modifications, and larger works may be distributed under different terms and without source code.
Copyleft
Apache License 2.0
Contributors provide an express grant of patent rights. Licensed works, modifications, and larger works may be distributed under different terms and without source code.
GNU Family
You are allowed to use, redistribute and change the software, but any changes you make must also be licensed under the GPL with different flavors.
GNU GPLv3
Permissions of this strong license are conditioned on making available complete source code of licensed works and modifications, which include larger works using a licensed work, under the same license. Copyright and license notices must be preserved. Contributors provide an express grant of patent rights. Same license required.
GNU LGPL v3
Permissions of this copyleft license are conditioned on making available complete source code of licensed works and modifications under the same license or the GNU GPLv3. Copyright and license notices must be preserved. Contributors provide an express grant of patent rights. However, a larger work using the licensed work through interfaces provided by the licensed work may be distributed under different terms and without source code for the larger work.
GNU AGPLv3
Permissions of this strongest copyleft license are conditioned on making available complete source code of licensed works and modifications, which include larger works using a licensed work, under the same license. Copyright and license notices must be preserved. Contributors provide an express grant of patent rights. When a modified version is used to provide a service over a network, the complete source code of the modified version must be made available. Requires same license, statement of changes
MIT LICENSE
MIT License Copyright (c) [year] [fullname] Permission is hereby granted, free of charge, to any person obtaining a copy of this software and associated documentation files (the "Software"), to deal in the Software without restriction, including without limitation the rights to use, copy, modify, merge, publish, distribute, sublicense, and/or sell copies of the Software, and to permit persons to whom the Software is furnished to do so, subject to the following conditions: The above copyright notice and this permission notice shall be included in all copies or substantial portions of the Software. THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
What is high performance computing in the context of bioinformatics?
Biomedical informatics involves big data. One of the biggest drivers lately is genomics and sequencing. Lets go with that example. Aligning a genome is a problem that is easy to break up. Aligning 1 read doesn’t impact aligning another read. If I have 1 billion reads, and a single CPU can align 2500 reads per second it will take 72 hours. If you want the answer quicker you need to split the analysis up and send it off to different computers that ideally all share network storage so that we can keep track of data. This is solved by High-Performance-Computing.
At this point, we should highlight there are three major areas of HPC that bioinformatics sees:
(1) Problems that can be easily broken up – divide and conquer. Analysis of genomes is a great example. Divide and conquer works well when the core calculation doesn’t require knowing what the result is from other calculations. For example, aligning 1 read to a genome, frequently (except for assembly), aligns to the same position in the genome regardless of what everyother read aligns. This is true at least at early steps, and thus we can take a very large file of reads of data and split up the work such that each computer is told to work on a subset of reads. In some programs this can lead to several independent alignment files or BAMs, that must be put together and sorted. Sorting of course would require each read knowing the location of all others – fortunately it is fast enough not to require splitting up across nodes. We will see this by example later.
(2) Problems that require lots of communication at each step & are less easy to break up. Molecular modeling and dynamics is the most relevant example where the positions and velocity of atoms are known, the forces interacting between them are known based on their location, and one wishes to move them all an infinitesimal small step forward based on this info, then recalculate the forces once the atoms have moved. In this scenario, we need to know where all the atoms (x,y, z in cartesian space) are and their current velocity (v), calculate where they will be 1 picosecond later by integrating Force=mass*acceleration, where acceleration is dv/dt. Millions of atoms interacting may take up to 1s across dozens of computers to calculate, but they all need to report back their updated position to calculate the next picosecond. That requires lots of communication at every second since each ps is typically 1 to 500 milliseconds, and this is done often using MPI (Message Passing Interface). Example of titin unfolding visualized in VMD, but run with NAMD. NAMD is the package which does the MPI molecular dynamics.

This example from 1997 is simulation of Atomic Force Microscopy pulling on a single protein domain. It took 200,000 CPU hours on Origin 2000 SGI box (imagine Pentiums in big shiny glowng box), generating 200Gbytes of data. It taught the field about how mechanical stablity – particularly relevant in cell-adhesion, wound recovery, and cell migration was impacted by protein structure. Peter Coleman and Martin Karplus helped pioneer the framework of how we modelled and simulated proteins, which of course led to tremendous insights into rational drug design using computational modelling. This type of computing required heave MPI code. With the nobel going to mind-numbing innovation in our ability to resolve protein structure using Cryo-EM, these types of analysis may have new life after largely being under the radar for the last 15 years.
(3) Problems that be done on GPUs (rarely relevant and development area). Very few applications today use GPUs. These are basically video cards where each one has 1000s of processors but with very little memory. They are largely an area of development and very little to any routine analysis is done on them for applied biomedical informatics at this point.
Lets focus on application #1 – analysis of genomes. Lets just consider the size to help us understand why your laptop may not be sufficient and you need industrial sized computing.
First, how much data is typical for 1 person’s genome?
First lets think about the data size and get perspective. The raw data for sequencing a person genome at 30x is 100 billion bases, where each base has a call and a quality measure of that call. Typically, 100Gb or more. 30x sequencing of a 3 billion base genome produces a lot of data. Lets talk about analysis starting at raw-data level. Typically, we sequence a small fragment of DNA – perhaps a 300 base segment – reading the first 100 bases and last 100 bases. For the germline (or inherited genome) above that is 1 billion reads. The first step is usually aligning those sequences to the reference genome. Remember we aren’t just aligning we are aligning data that will be slightly different. The person we sequencing will have variants, and thus we have to allow mismatches, insertions and delewtions. Moreover, there will be error. The reads aren’t perfect but about 99.5% accurate meaning that typically there are 1 to 2 machine errors per read. As a result, its good to sequence multiple times because one typically wants to get to six sigma – or 1 in 1 million accuracy. So if you have 100 or so individuals, and you have 10 terabytes on your hand.
Bioinformatics is big data – but where in the big context?
- The simplest unit is Boolean – 0 or 1 and is a bit.
- A character is a single byte – “A” is one byte and typically 8 bits
- An email contains thousands of bytes
- The Nintendo 64 had 4096 bytes of memory
- The C64 had 64,000 bytes
- The first 5.25″ floppies were 640,000 bytes
- The smaller 3.5″ had typically 1-3 Mbytes
- An MP3 is 3 to 4 Mbytes.
- A microarray often produces 60 Mbytes of data (bioinformatician hour)
- A CD-rom stores 600 million bytes;
- A DVD is 6 billion bytes or gigabytes (GB)
- Flash USB storage can hold 8 to 64 Gbytes
- A 30x genome is 100-300 billion bytes for the BAMs and FASTQs(bioinformatician week)
- Portable hardrives have about 2 trillion bytes (2 Terabytes or 2TB)
- 100 individuals ($100K in reagents) requires 30 trillion bytes (30 Terabytes)(bioinformatician year)
- Amount of data indexed by Google monthly is 200 Terabytes by approximately 30,000 software engineers
- 1 NovaSeq 6000 will produce 365 Terabytes per year at full capacity (2 to 3 bioinformatician )
- CERN study of “God particle” for Haldron Collider generates 5 Petabytes/yr (1000 Terabytes)
- All the internet data 8000 Petabytes
So its not unusual for a lonely bioinformatician or two to be analyzing and managing data sizes (10-200 TB) that are an order or two in magnitude away from what is largely considered the most data rich scientific activity – the search for God particles through the CERN by 1000s of scientists, or what we often think of as the internet (the 0.004% that is being indexed by Google). Hundreds of terabytes is a lot of data.
So what does in terms of processing power, let alone storage to align 1 billion reads in your 30x genome? BWA is the name of a very fast and accurate aligner. It can align 2500 reads per second, and it would take about 75 hours. What if you want it faster? Well we could split it up and have a few computers work on alignment. This frames High Performance Computing (HPC) for buioinformatics. Speaking broadly first, HPC is typically an environment with lots of “computers” or rather “compute nodes” all networked together with lots of bandwidth between them, enough memory to run the calculation, and data storage to put the data.
That much data, we need to think about how long even moving it will take.
How long does it take to transfer 1 person’s whole-genome data?
3G/4G ~ 70 hours
Cable Modem (50 Mbit) ~ 30 hours
USB – 1 to 3 hours with standard USB, though newer USB is Gigabit+.
T3 University to Amazon ~ 1 to 3 hours (some parallel and high-end transfer, but you are saturating tyupical ports).
Pragmatically, its hard to move faster than 400 Mbit to 800 Mbit for a given file across the internet
Key HPC Concepts
More exactly HPC has a few important concepts bioinformatics must know well, and we start by providing a key question driving why that concept matters.
(1) Compute nodes.
Example Question: How many computers and how many cores do they have?
(2) Bandwidth (i/o)
Example Questions: How long does it take to read and write data, particularly if lots of processors or writing and reading on shared storage? How do we get the data into the HPC system?
(3) Memory
Example Question: How much do we need to put into memory – surely, a 3 Gbyte human genome is difficult to put into 2Gb of memory.
(4) Storage.
Example Question: How much storage do we need – where is it and how is it maintained and managed. Where do we put data before and after.
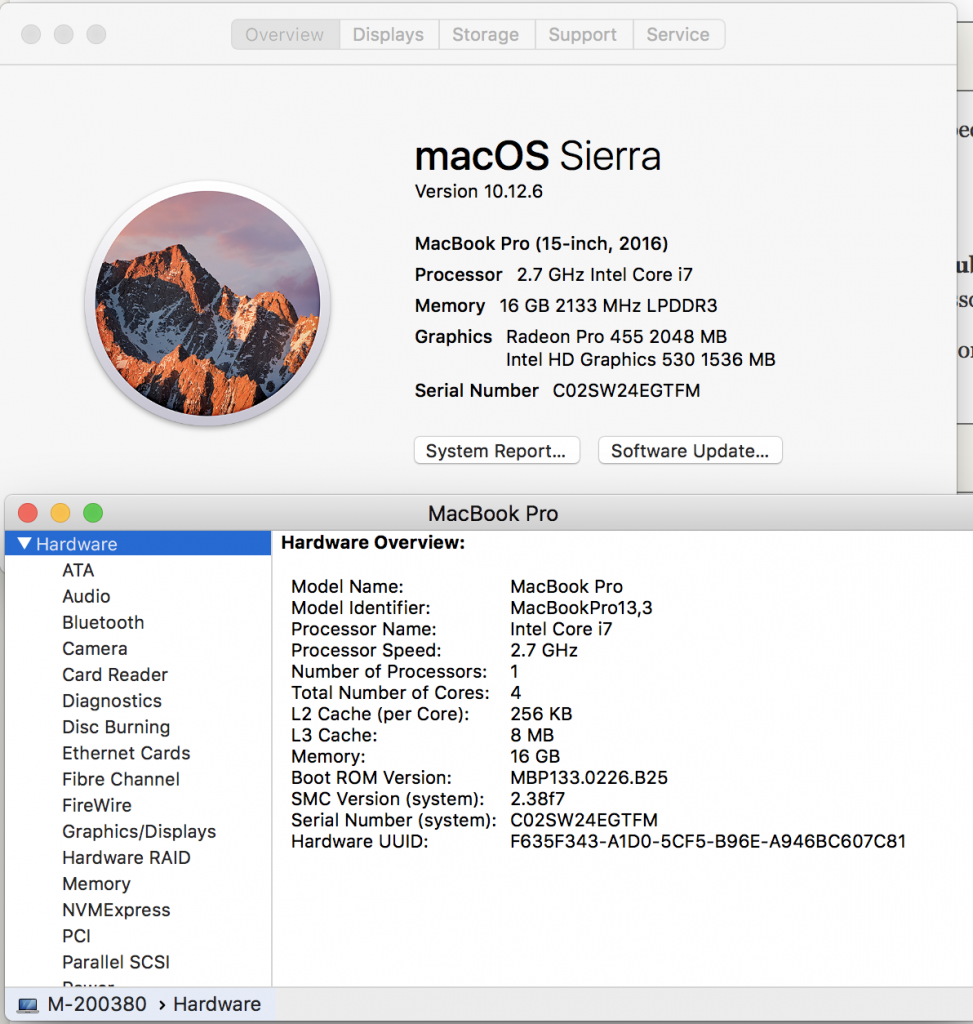
Lets learn about each of these by understanding your personal laptop. In the upper left hand corner of your screen press the apple icon, and select “About this Mac”. We see information about processors, Memory, and Graphics. Lets go deeper by clicking on System Report and look at Hardware Overview.
Your laptop is like a compute node. We can see in the example below this laptop has an Intel Coreil at 2.7Ghz with 1 processors and that processor has 4 cores. Each core can handle a thread. A thread would be for example a script (noting you can always spin off more process within your script using &, and sending to background). We have 16 Gb of memory with 8Mb of L3 Cache and 256 of L2 Cache. These latter two are important, but the key for us is the 16 Gb. The more memory the more things we can open at once – more windows, etc.
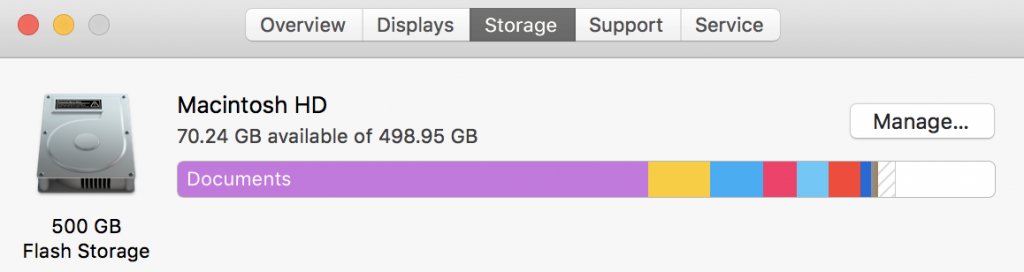
We can also click on storage from the “About our Mac” and see how much storage we have and how much is being used. Storage isn’t active – its like having a lot of music on a portal MP3 player – except MP3s take up a little bit space. In this case I have 500 Gb of flash storage. Flash storage is fixed and doesn’t involve a spinning disk – your new iPhone and your iPads are flash, and the original large iPods were spinning disks. Spinning disks usually hold more – they also and always eventually break.
Data Management: Digging deeper
Everything that is glass will probably break, and all disks will fail. Data is expensive to create, and a bioinformaticians job is to often maange data. Losing data can and is a massive problem – it can cost millions, it can cost time, it can get people fired, and with certain types of health data you can be prosecuted. Its a bioinformaticians job to work with IT and scientists to make sure that the risks are known and what the current strategies are. If data isn’t backed up, it is a roll of a dice typically of when that data will be irrecoverably lost. Documentation is key, and processes are key.
Critical data should be backed up in two geographically different locations. Fortunately we live in the era of cloud computing – that is on demand storage managed by IT professionals available in $0.007 increments (store 1 Gbyte for 1 month, and you get charged a fraction of a cent at Amazon). I’ve seen companies get shut down because of water sprinklers triggering a fire. In this sense data management costs significant amount of dollars, and done poorly the expense can be massive.
Lets dive into a High Performance Computing (HPC) environment. In HPC, there is local storage and various networked storage. Network storage is usually not on your computer and is somewhere on the network. That means if its say at a location with just a little bandwidth, it’ll be slow to load on your computer. There are typically hundreds to thousands of CPUs reading from this storage, and they very easily can use up bandwidth. Thus there are a different types of storage and we do need to know more about them in bioinformatics. Now its important to know that these are rarely a single drive, but rather many many drives where data from your project is written across them somewhat redundantly. RAID storage is this – your data is striped across a bunch of drives and provides two advantages. First it helps spread out the bandwidth so that you can get simultaneously read across lots of drives. Second, if a drive files there is typically enough redundancy that you are fine and your data isn’t lost.
Critical network storage servers types
Each HPC environment has their own setup, but there is convention – and frequently common themes about which data storage is meant for calculation and temporary storage and which data storage is for long-term storage, etc. On any computer you can always see what’s mounted by ‘df -h’. The list is long, and only a few are really critical – scratch, long-term storage, and your home directory. These are often served to your computer on different servers, and then mounted made available by a simple ‘cd /yourServer‘. Alias’s and links can sometimes mask when you switch storage – so one must be careful so as to not run out of space. This frequently occurs by newbies or new users storing all their data in their home directory. Often this space is small -purposely so – to keep you from hoarding data where you shouldn’t.
Stratch or Staging Storage. This is typically large continuous data. Its often optimized to handle lots of computers reading and writing tons of data simultaneously. Its also met to be temporary storage, so there is less redundancy. Often when we are doing an analysis we will read and write to a scratch storage.
df -h | grep staging beegfs_nodev 328T 312T 16T 96% /staging
Long-term storage. Staging is another continuous block of data storage. It is optimized for you to upload your datafiles once, and your data is safe there for short-periods of times.
ln -s/auto/dtg-scratch-01/dwc3/davidwcr/ ~/storage #To get to our storage, thus cd ~/storage
Home storage. This is the storage that holds your home directory and some of the files and programs you use. Its usually not that big, and you shouldn’t really upload large datasets.
davidwcr@hpc-login3:~ df -h | grep rcf-40 almaak-05:/export/samfs-rcf/rcf-40 5.5T 2.1T 3.5T 37% /auto/rcf-40
Other types of data storage
Glacial storage. This is a long-term back-up option where you don’t need data but very rarely, and its effectively written to a storage type that can take hours to retrieve, but the cost of storage is very cheap
Cloud storage. This is storage typically at a commercial vendor such as Google or Amazon – but under the hood the storage is typically not that different from some of the other storage types above, other than we need a networked connection to Google.
Local storage. Each node or computer usually has a small amount of storage built in – typically from a hard-drive and sometimes you want to use it for data that needs to move back and forth a lot between disk and memory.
Tape storage. This is usually for long-term backup, where it can take hours to weeks to get data from tape. These literally are tapes, and sometimes its robots that go and get the right tape and sometimes its humans.
There are often many many other types of storage. One way to see all the storage available is to login and type “df -h”.
Data needs to be organized. Primary data should be in an logical order and hierarchal